|
|
Dr. F. Parodi - Industrial R&D Expert Expertise & Know-How: Computational Chemistry & Modeling |
|
|
Dr. F. Parodi - Industrial R&D Expert Expertise & Know-How: Computational Chemistry & Modeling |
| computational chemistry & modeling | | chemical analysis | | consulting & advisory | |
_____________________________________________________
Computational Chemistry & Molecular Modeling
|
Calculation of Physical and Chemical Properties of Original & Novel Organic Compounds |
|||
|
Computation of molecular properties of novel, not yet or not-enough investigated organic compounds, as well as unstable or inaccessible reaction intermediates, in the gas phase or in solution, at any desired temperature:
|
|
||
|
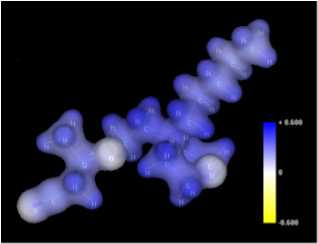
Analysis of molecular properties under the influence of electric and/or magnetic fields with predetermined direction and intensity; calculations of molecular polarizability (mean value and tensor components). Building and analysis of polymeric and crystal structures. |
|||
|
Usage of a range of computational chemistry methods:
Properties computed for predetermined molecular conformations. Average, equilibrium values obtained via molecular dynamics or Monte Carlo simulations. Compounds in solution investigated via Langevin dynamics simulations. |
|||
| Redesign & Optimization of Chemical Reactions & Reaction Catalysts | |||
|
Computer-aided, quantum-mechanical modeling of organic reactions:
|
||
|
|||
|
Calculation of Physical and Chemical Properties of Original & Novel Polymeric Substances |
|||
|
|
||
|
|||
|
|||
|
|||
__________________________
|
Molecular simulations and data processing carried out on Intel i7 ® workstations [under Windows 7 (64-bit) ® operating system (Microsoft Corp.)] |
back to index: "Expertise & Know-How"
| computational chemistry & modeling | | chemical analysis | | consulting & advisory | |
|
© Dr. Fabrizio Parodi - 2000-2015 |