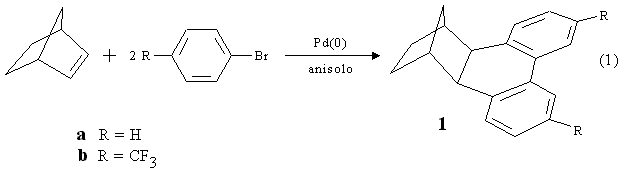
La funzionalizzazione di nuclei aromatici è un importante obiettivo della chimica ed in particolare della chimica organica industriale. I metodi di sintesi messi a punto in tale direzione nei nostri laboratori, ci hanno permesso di ottenere in modo catalitico strutture aromatiche di vario tipo attraverso reazioni "one-pot" molto semplici dal punto di vista preparativo. Ha dato inizio a questo tipo di lavoro la sintesi dell'esaidrometanotrifenilene 1 a partire dal norbornene e bromobenzene in presenza di Pd con t-BuOK come base in anisolo (eq. 1) [1].
Per fare luce sul meccanismo di tale reazione furono isolati diversi intermedi organometallici. Nel presente lavoro si è seguito il criterio di intrappolare gli intermedi di reazione mediante olefine per ottenere indicazioni inequivocabili sull'andamento della reazione. Così facendo si è anche arricchita la chimica della funzionalizzazione aromatica catalizzata dal palladio con nuove realizzazioni sintetiche.
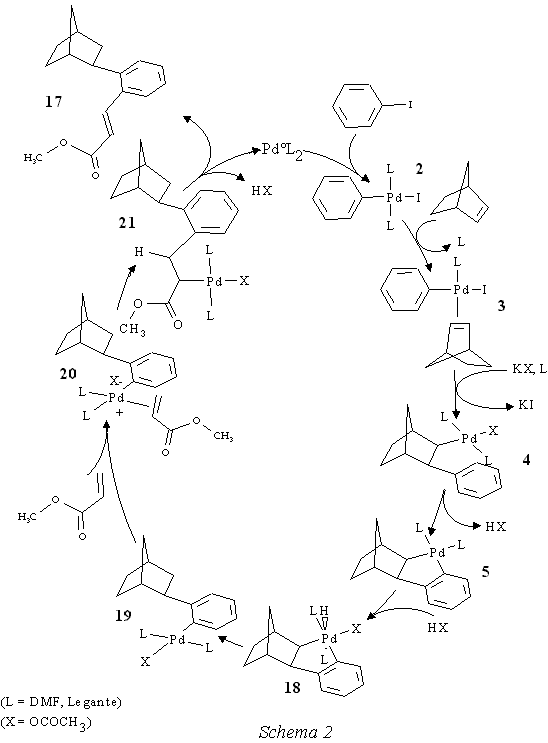
Si riassumono nelle pagine che seguono gli studi effettuati nei nostri laboratori ed all'estero sull'andamento della funzionalizzazione aromatica catalizzata da metallacicli alchilaromatici del palladio. Gli studi rivolti alla comprensione del decorso di reazione della reazione (1) hanno consentito d'isolare gli intermedi organometallici 4-7. L'andamento della reazione (1), esemplificato nelle equazioni successive per il legante PPh3, implica la formazione del prodotto di addizione ossidativa dello iodobenzene al Pd0 2 seguita dall'inserimento del norbornene per dare 4 (facilitata dallo scambio con un anione X meno coordinante come CH3 COO- o HCO3 - [2]) (eq. 2).
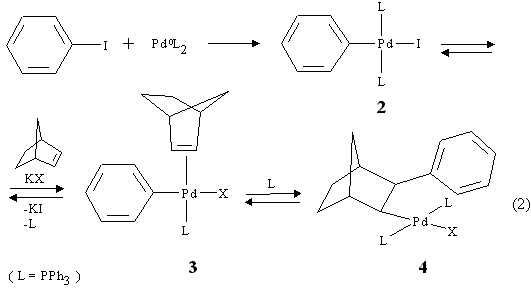
Segue la formazione del palladaciclo 5 (eq. 3). Questo complesso ha geometria cis ed exo in quanto sia il fenile che il Pd sono rivolti dallo stesso lato rispetto al norbornano ma anche dalla stessa parte in cui si trova il gruppo metilenico a ponte. L'eliminazione del Pd e di un idrogeno in è sfavorita poiché essi sono tra loro in posizione anti su una struttura rigida [3].
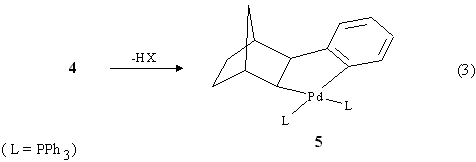
Nel successivo passaggio si ha l'addizione ossidativa dello iodobenzene al complesso di PdII con formazione di una specie di Pd IV 6(eq. 4):
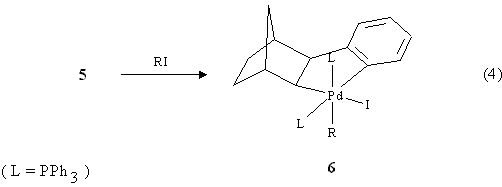
L'intermedio 6 dovrebbe contenere R = Ph in quanto la sua ulteriore evoluzione dovrebbe portare all'esaidrometanotrifenilene ma è stato possibile isolarlo solo quando R = alchile. Per eliminazione riduttiva da 6 si forma il complesso di Pd II7 (eq. 5):
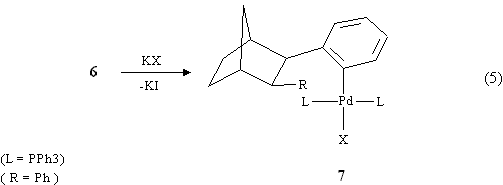
Infine per successiva sostituzione elettrofila intramolecolare si ottiene l'esaidrometanotrifenilene 1 e contemporaneamente si rigenera la specie di Pd 0 che può iniziare un altro ciclo catalitico (eq. 6).
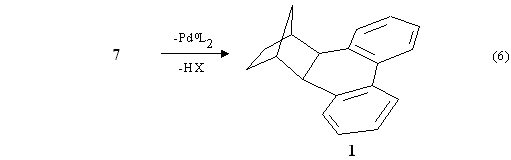
Un interessante sviluppo delle reazioni studiate nei nostri laboratori è dovuto ad un gruppo tedesco (de Meijere) [4] che lavorando in condizioni leggermente diverse ha ottenuto un esaidrometanotrifenilene contenente un fenile in più8 (eq. 7).
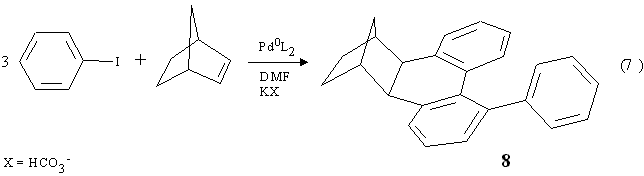
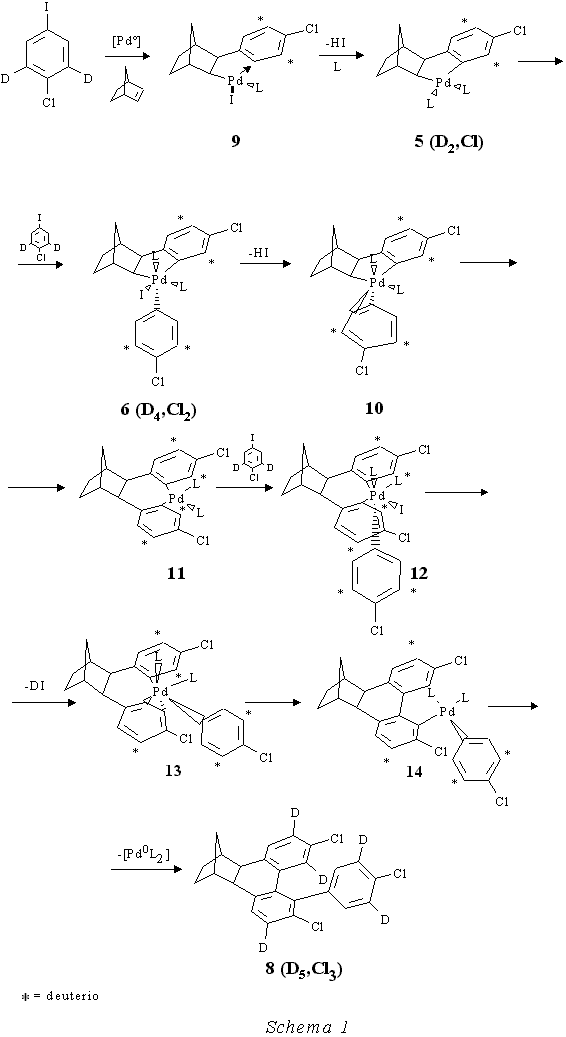
Nel meccanismo da loro proposto (Schema 1 a pag. 7) per spiegare il prodotto 8 essi ipotizzano, come nel caso dell'esaidrometanotrifenilene 1, il passaggio attraverso un complesso arilico di Pd IV 6 (D 4,Cl 2) che però per eliminazione di acido alogenidrico si trasformerebbe in un complesso arinico di Pd IV 10. Quest'ultimo reagirebbe subito dando un'eliminazione riduttiva per formare un palladaciclo a 7 termini 11. Questo sarebbe in grado di dare una terza addizione ossidativa formando 12 ed in una sequenza di stadi di eliminazione di acido alogenidrico in cui si riforma l'arino 13 e di due successive eliminazioni riduttive con formazione di legami C-C attraverso complessi di PdIV e PdII, darebbe il prodotto 8 (D5 ,Cl3) e la specie di Pd0cataliticamente attiva. Il meccanismo indicato si presta tuttavia a varie critiche come si vedrà più avanti. Con l'intenzione di capire quali fossero gli stadi reattivi realmente implicati nella formazione di 8 abbiamo bloccato con olefine gli intermedi coinvolti nella reazione (eq. 7). I risultati ottenuti hanno permesso di arrivare ad un'interpretazione alternativa a quella di de Meijere che rende conto di tutti i fatti osservati e consente inoltre un ulteriore avanzamento delle metodiche di funzionalizzazione aromatica.
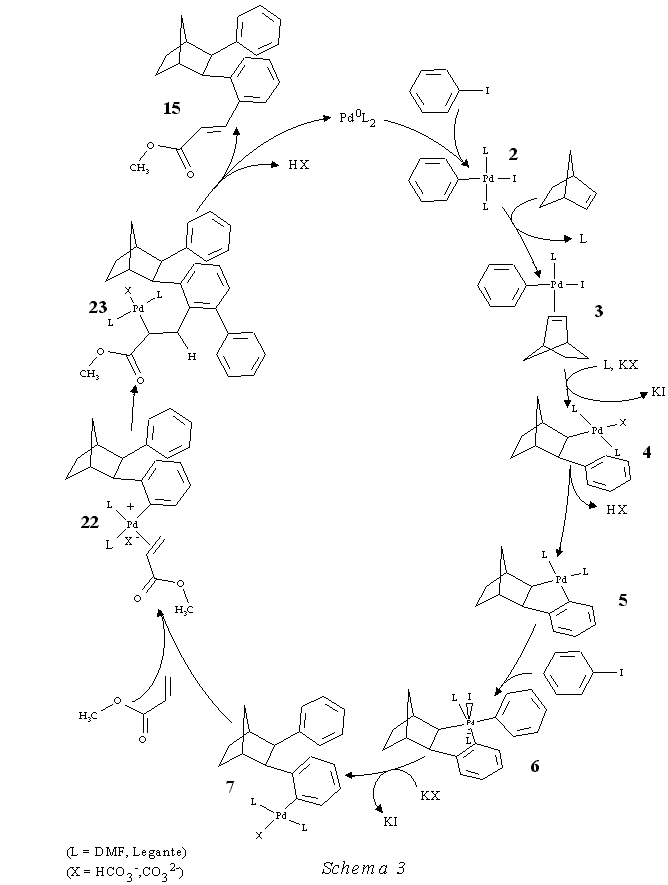
Facendo reagire iodobenzene, norbornene e acrilato di metile in presenza di Pd(CH3COO) 2 e di K 2CO3 in dimetilformammide a 80°C per 18 ore si sono ottenuti come prodotti principali i composti 15a e 16 rispettivamente con rese gascromatografiche superiori al 10% e al 20% accanto a numerosi altri prodotti presenti in piccola percentuale. Introducendo nello iodobenzene un sostituente CF3 in para la reazione condotta nelle stesse condizioni ha portato alla formazione di 15b con resa in prodotto isolato del 10% e a 1b con resa del 9%.
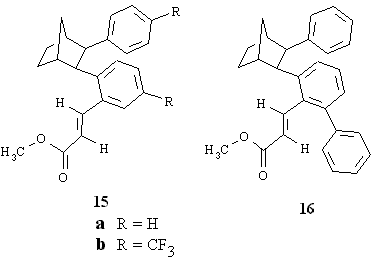
Noi abbiamo rivolto l'attenzione ai prodotti 15 e 16 perché la loro presenza ha significato per l'interpretazione del corso delle nuove reazioni studiate. La stessa reazione di sopra condotta a 105°C per 18h, ma utilizzando come base CH3COOK al posto di K2CO 3, ha portato alla formazione di 17 e di un altro composto non ancora ben caratterizzato di massa 350, la cui struttura è simile a quella di 17 ma con un'unità norbornanica in più. Inoltre è presente il cinnamato di metile che deriva dalla nota reazione di addizione di fenile all'acrilato di metile [5].
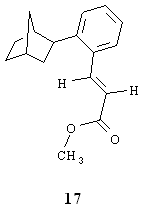
Di speciale interesse risulta essere il composto 16 in quanto non spiegabile nè secondo gli intermedi implicati nella sintesi dell'esaidrometanotrifenilene nè secondo il meccanismo proposto dal gruppo di ricerca tedesco quale riportato nella parte introduttiva. I prodotti 15, 17 e probabilmente anche quello di massa 350, che si omette dalla discussione perché non ancora completamente caratterizzato, si possono spiegare facendo riferimento agli intermedi isolati per chiarire l'andamento della reazione dell'esaidrometanotrifenilene. La formazione di 15 e 17 inizia con l'addizione ossidativa dello iodobenzene alla specie cataliticamente attiva di Pd0. Nel passaggio successivo si ha la coordinazione ed inserimento del norbornene nel legame arile-Pd prima che lo possa fare l'acrilato di metile. Anche se quest'ultimo è presente in forte eccesso, il norbornene, essendo un'olefina rigida e tensionata, ha proprietà coordinanti notevolmente migliori dell'acrilato di metile e si coordina per primo al Pd. Successivamente per eliminazione di acido si ha la formazione del palladaciclo 5. A questo punto nel caso di 17 (reazione con CH3 COOK come base) si passa, attraverso addizione ossidativa dell'acido acetico presente al PdII, ad una specie idrurica di PdIV 18, che si trasforma spontaneamente in un complesso di Pd II in cui il palladio è legato all'arile 19. L'olefina (acrilato di metile) si coordina al complesso di Pd IIdando 20 per poi inserire nel legame Pd-arile per formare 21. Per eliminazione riduttiva si rigenera la specie attiva di Pd0che può iniziare un nuovo ciclo catalitico mentre contemporaneamente viene rilasciato 17 e acido acetico secondo lo Schema 2 a pag. 11.In questo come negli schemi successivi gli stadi in cui avviene lo scambio dello ione ioduro con l'anione X dell'acido debole sono indicati "tentativamente".
Nel caso di 15 (per semplicità si fa riferimento a 15a nello Schema 3 a pag. 12), che si ottiene in presenza di K 2CO 3 come base, dopo la formazione del palladaciclo 5 una nuova molecola di iodobenzene (anziché l'acido acetico che interviene nella sintesi di 17) dà l'addizione ossidativa al Pd II, con formazione di 6. La successiva migrazione dell'aromatico sull'alchile conduce a 7, dopodiché il metallo coordina l'acrilato di metile che inserisce sull'arile per dare 23. Per eliminazione riduttiva dell'acido si forma 15 e Pd 0L 2 che ricomincia un altro ciclo catalitico (Schema 3).
Risultati analoghi si hanno usando un'olefina come lo stirene al posto dell'acrilato di metile. Infatti facendo reagire norbornene, iodobenzene e stirene in presenza di Pd(CH3 COO)2 con K 2CO 3 in dimetilformammide per 18 ore ad 80°C si sono ottenuti come prodotti principali 25 con resa gascromatografica del 24% e 24 (la struttura di quest'ultimo è ancora da confermare) con resa del 12%.
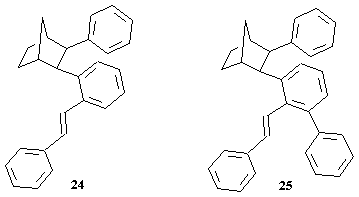
Mentre il prodotto 24 potrebbe essere ben spiegato seguendo un decorso analogo a quello di 15, la formazione del prodotto 25 in analogia a quella del prodotto 16 presenta degli aspetti che devono far supporre che essi derivino da un altro tipo di ciclo catalitico. Il cammino che porta a 16 o 25 è comune con quello che porta a 15 fino alla formazione di 7 (Schema 3 a pag. 12). Tale intermedio a questo punto inserisce l'olefina dando 15 o 24. Per spiegare la formazione di 16 o 25 occorre rendere conto del perché si verifichi l'attacco di un'arile in posizione orto al legame col palladio dell'intermedio 7 considerato . Abbiamo per questo avanzato un'ipotesi meccanicistica che prende spunto dall'interessante comportamento di complessi di arilnorbornilpalladio(II) orto, orto-disostituito [6]. Tali complessi, la cui struttura corrisponde a 30, in cui il nobornile legato al palladio si viene a trovare in condizioni di forte compressione sterica da parte dei sostituenti in posizione orto sull'arile, tendono a decomporsi spontaneamente per dare il complesso arilico di palladio(II) del tipo 31 e norbornene.
Si è quindi pensato che il norbornene potesse svolgere un ruolo analogo nella formazione di 16 e 25 secondo il meccanismo qui sotto indicato (Schema 4 a pag. 15). Il punto più discutibile di questo meccanismo riguarda il perché il terzo fenile entri sul fenile e non sul norbornile del metallaciclo 29 in contrasto con quanto precedentemente osservato da noi. Infatti in precedenti nostri lavori era stato messo in evidenza come regola generale che se è un arile a dare l'addizione ossidativa ad un palladaciclo con formazione del complesso 6 l'eliminazione riduttiva successiva lo porta invariabilmente sulla parte alchilica della struttura legata al Pd (7, R = fenile) [7]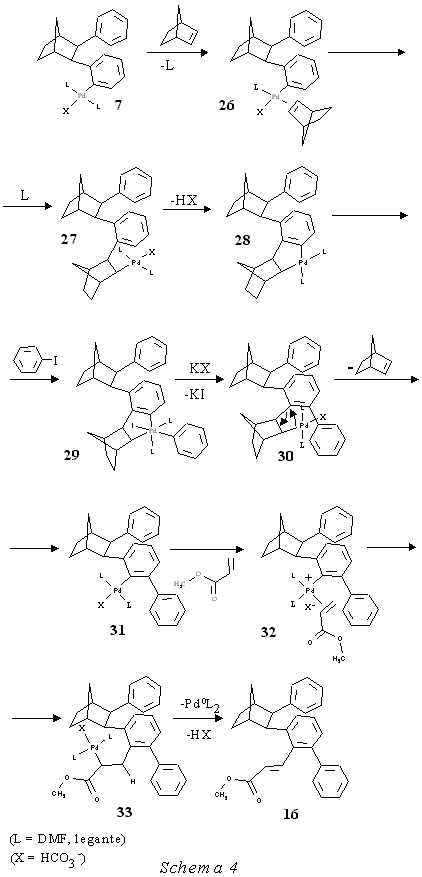
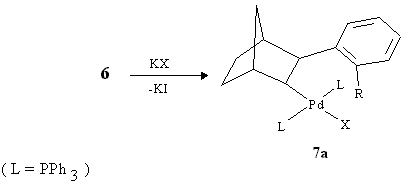
Se invece R è un alchile esso va sempre sulla parte aromatica del complesso (7a).
E' tuttavia da osservare che da una prova effettuata con 4-bromo-trifluorotoluene e 2-norbornene in presenza di palladio acetato e di carbonato di potassio in dimetilformammide, si è ottenuto un prodotto 34 che corrisponde alla migrazione dell'arile sull'arile con resa in prodotto isolato del 15% (assieme al prodotto chiuso 1b con resa del 60%).
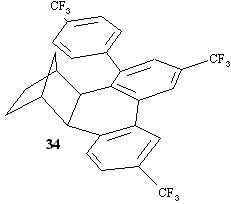
Anche se si tratta di un risultato limitato al gruppo CF 3, esso indica comunque che la migrazione di un'arile su un'arile è fattibile in particolari condizioni. La spiegazione del diverso comportamento che si verificherebbe a livello del metallaciclo del Pd IV29, riportato nel precedente Schema 4, può trovarsi in un decorso concertato in cui il fenile che entra provoca in concomitanza l'allentamento della tensione sterica.
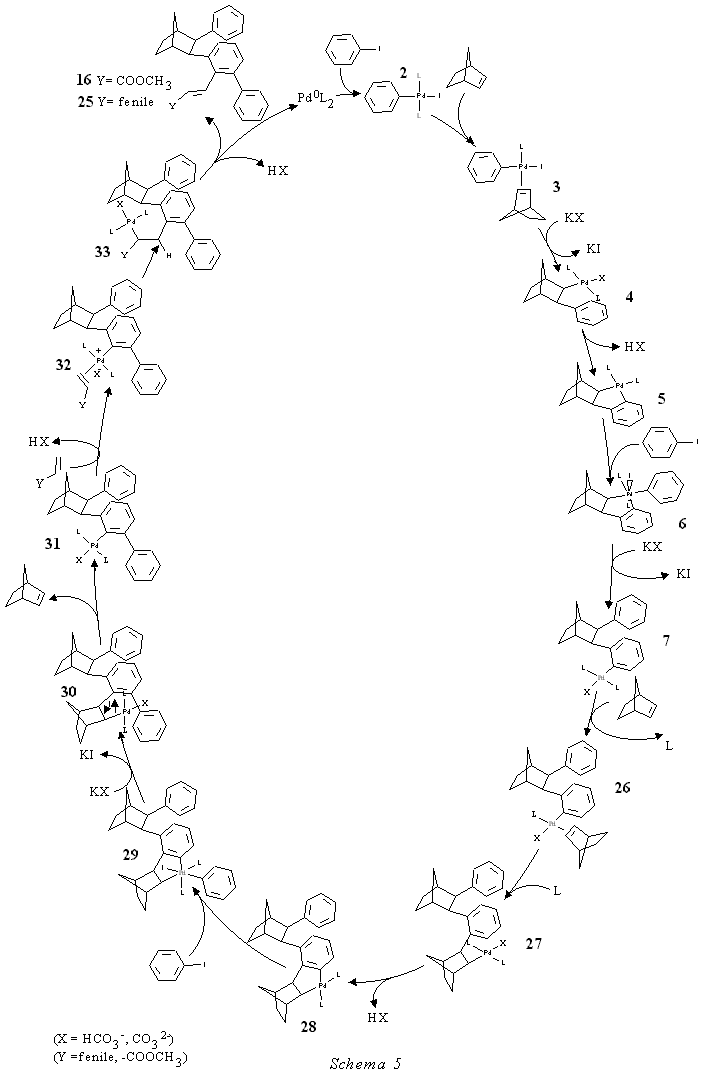
E' da rilevare che il suddetto ciclo sia il solo che può rendere conto in modo completo dei prodotti isolati. A questo proposito abbiamo anche tentato di applicare al nostro caso il meccanismo arinico postulato da de Meijere. Lo schema 6 mostra che, mentre i prodotti più probabili dovrebbero essere gli ultimi tre 37, 38 e 40 corrispondenti il primo all'attacco dell'olefina all'arene meno impedito e gli altri due all'espansione del metallaciclo [8], e il prodotto isolato 16 male si concilia col meccanismo proposto in analogia a quello di de Meijere.
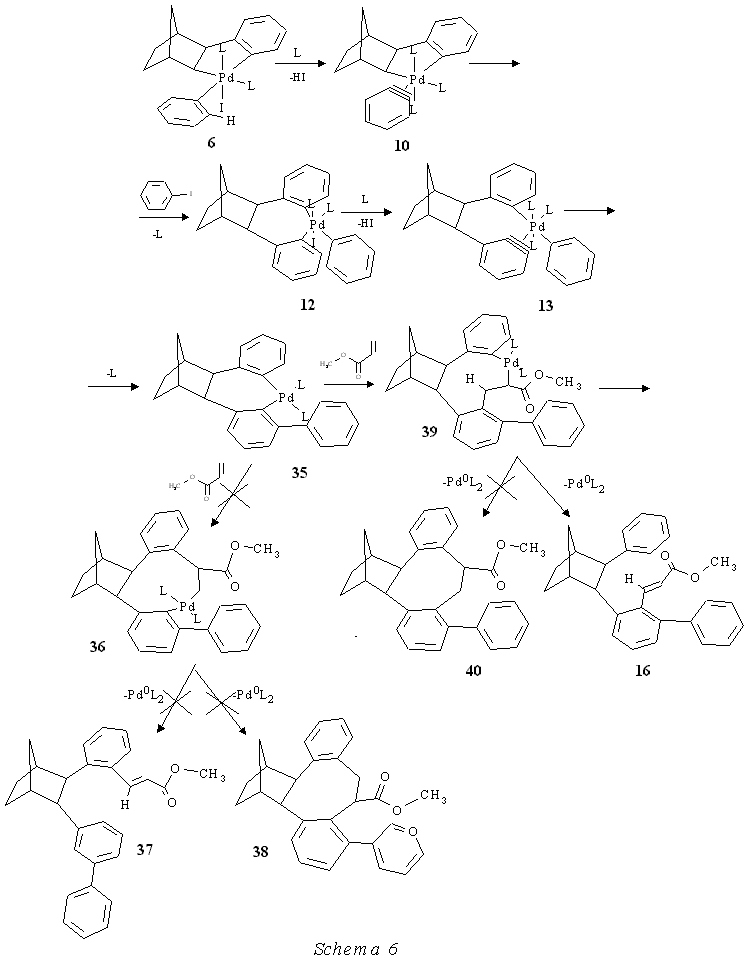
In considerazione del fatto che il meccanismo da noi proposto giustifica in modo soddisfacente la formazione dei prodotti ottenuti in presenza di olefine ci siamo domandati se esso non fosse anche il più adatto per rendere conto in modo razionale del meccanismo della reazione descritta da de Meijere che conduce al composto 8. Riferendoci allo Schema 1 riportato nell'introduzione a pag. 7 dobbiamo osservare che il meccanismo arinico proposto da de Meijere mostrava alcuni punti non chiari. Infatti l'intermedio 10 dovrebbe dare oltre che 11 anche l'isomero con il cloro in posizione para rispetto al legame col palladio del metallaciclo 11a (derivante dall'attacco del norbornile all'altro carbonio arinico) mentre invece il prodotto corrispondente 8a (D5, Cl3) non si forma (Schema 7).
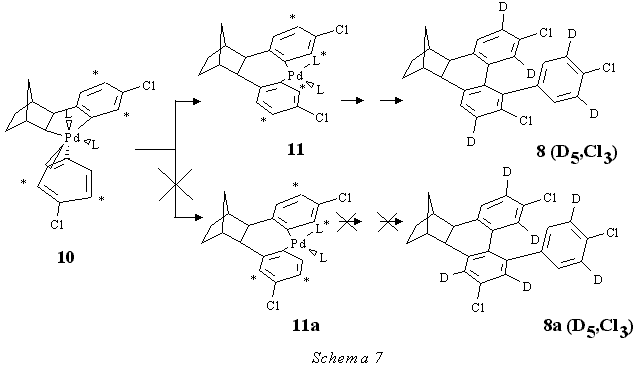
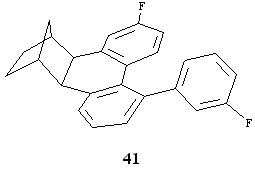
La possibilità che un metallo di transizione possa stabilizzare un benzino [9] fa riferimento a lavori in cui si sono studiati complessi di metalli di transizione di cicloalchini e arini. In tale lavoro si riporta che questi arini, che in altre condizioni sono intermedi tanto reattivi da non potere essere isolati liberi, possono essere stabilizzati da complessi di Ni o di Pt contenenti fosfine. In tale lavoro in particolare si mettono in risalto le condizioni fortemente riducenti utilizzate per la formazione dell'arino, che sono molto diverse da quelle in cui noi e i ricercatori tedeschi operiamo. Infatti il complesso mononucleare benzinico di nichel(0), ivi riportato, fu ottenuto per riduzione del complesso(2-bromofenil)cloronichel(II) per mezzo di un'amalgama al 1% di sodio e solo per questa via.In maniera analoga a quelli di nichel furono preparati i complessi mononucleari benzinici di platino(0) PtL2 (C6 H4 ) (2L = 2PEt3 , 2PCy3 , Cy2 PCH2 CH2PCY2) (Cy = cicloesile) usando per la riduzione un'amalgama al 43% di sodio. I tentativi di preparare ML2 (C6H 4) (M = Ni, Pt; L = PMe3 , PPh3 ) attraverso il metodo riduttivo non ebbero successo. Altre precedenti nostre ricerche tese ad ottenere prove del coinvolgimento dell'intermedio arinico facenti uso del m-fluoroiodobenzene non hanno portato al prodotto 41 che risulterebbe dall'attesa formazione dell'arino dalla parte del legame F-C.
Per tutti questi motivi si è ipotizzato che il meccanismo di inserimento e disinserimento del norbornene si verificasse anche nelle reazioni che portano all'arilesaidrometanotrifenile-ne 8 secondo lo Schema 8 di pag. 22. Lo iodobenzene dà l'addizione ossidativa alla specie di Pd 0 formando un complesso di Pd II 2. Segue la coordinazione e l'inserimento del norbornene nel legame arile-Pd (3-4). Per eliminazione di HX si forma poi il palladaciclo 5 a cui una seconda molecola di iodobenzene dà l'addizione ossidativa formando una specie di Pd IV 6 che evolve dando l'intermedio 7. Una seconda molecola di norbornene si coordina ed inserisce nel legame metallo-arile. Per eliminazione di HX si forma il palladaciclo di Pd II 28 a cui una terza molecola di iodobenzene dà l'addizione ossidativa. Si ha a questo punto l'inserimento del fenile nel legame arile-Pd e la formazione di 30. Il complesso di arilnorbornilpalladio(II) orto, orto disostituito si decompone, a causa della tensione sterica generata dai sostituenti in orto, e dà il complesso arilico di palladio(II) 31 e il
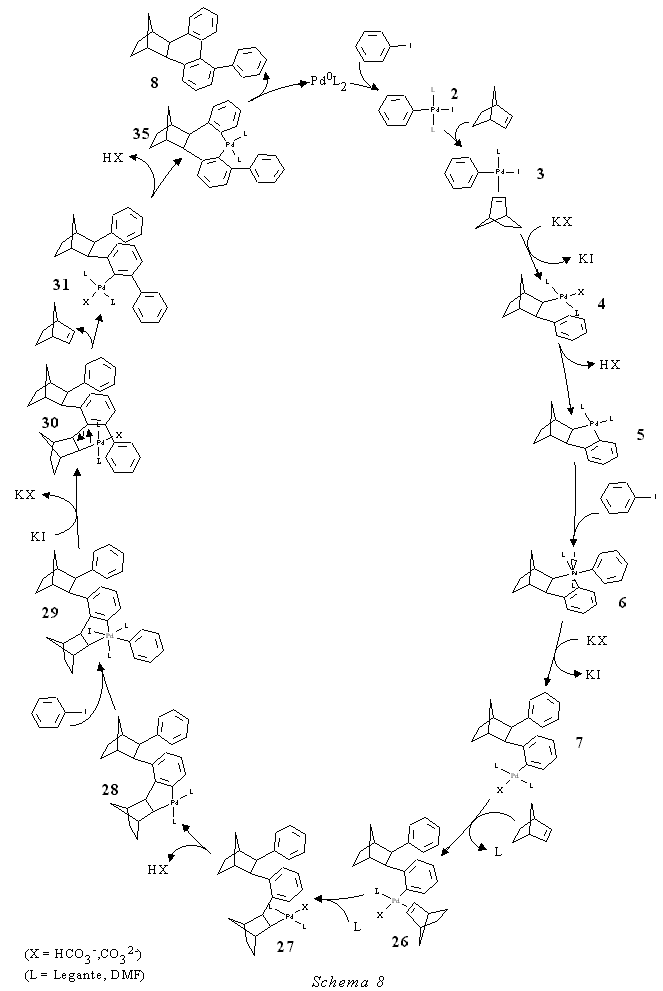
norbornene. Per successiva sostituzione elettrofila intramolecolare si ottiene l'arilesaidrometanotrifenilene 8 e Pd 0. Decisive per provarne la validità sono state le due seguenti reazioni. Nella prima reazione si è fatto reagire a temperatura ambiente per 24 ore in condizioni stechiometriche il complesso di fenilnorbornilpalladio(II) cloruro (4, X = Cl) e lo iodobenzene (rapporto molare 1:2) in presenza di carbonato di potassio ed in assenza di norbornene. La reazione ha dato come prodotto principale l'esaidrometa-notrifenilene 1. E' importante rilevare la totale assenza del prodotto 8. La stessa reazione condotta in presenza di norbornene ha dato tra i prodotti principali l'arilesaidrometanotrifenilene 8. Il gascromatogramma ottenuto dalla gasmassa risulta essere una fotografia del percorso reattivo seguito nella reazione. Per un monitoraggio completo degli intermedi, queste due reazioni, che hanno chiaramente dimostrato la necessità della formazione di un nuovo metallaciclo con il norbornene per consentire la fenilazione in meta rispetto all'originario legame carbonio-alogeno, verranno ripetute sul complesso 7 (Schema 8, pag. 22) in corso di preparazione (vedasi parte sperimentale). Alla luce dei risultati ottenuti si spiega anche il comportamento osservato nella reazione del 4-bromo-trifluorotoluene con il norbornene in presenza di palladio acetato e di carbonato di potassio in dimetilformammide che dà luogo alla formazione del composto 34 ma non al prodotto descritto da de Meijere. Questo potrebbe essere spiegato in base all'effetto negativo (sterico ed elettronico) esercitato dal sostituente CF 3 sulla chiusura del metallaciclo che è il presupposto per la successiva fenilazione. Anche l'assenza dell'analogo del prodotto 16 contenente il sostituente trifluorometilico é giustificabile nello stesso modo. In conclusione si è potuto provare un nuovo meccanismo di funzionalizzazione aromatica selettiva assistito dalla formazione di un palladaciclo alchilaromatico. In pari tempo si è realizzato un nuovo tipo di sintesi consistente nell'attacco regioselettivo alle posizioni 1,2 e 3 di un nucleo aromatico.
Le analisi in gascromatografia sono state eseguite con GC DANI 3900 con colonna capillare OV 101 (metilsilicone) di 15 m avente diametro di 0.25 mm, dotato di rivelatore FID operante alla temperatura di 280°C. Le determinazioni quantitative condotte in gascromatografia sono state effettuate utilizzando standard interni e registrate con integratore HP 3394. Le analisi GC-GM sono state effettuate con un gascromatografo HP 5890 serie II con colonna SE (metilfenilsilicone) dotato di selettore di massa HP 5971 a quadrupolo. Gli spettri di massa sono stati registrati mediante uno strumento FINNINGAN 1020 con selettore di massa a quadrupolo. Per le separazioni cromatografiche flash si è usata una pompa DURAMAT cfg ProMinent e una colonna di dimensioni = 3 cm, h = 45 cm. Gli spettri di risonanza magnetica nucleare del protone sono stati eseguiti con strumenti BRUKER AC-300 e AMX-400, usando come solvente CDCl3 . I chemical shifts sono riportati in ppm riferiti al TMS ( = 0) usato come standard interno. Gli spettri di risonanza magnetica nucleare del carbonio sono stati eseguiti con lo strumento AC-300 BRUKER a 75.4 MHz. I chemical shifts sono riportati in ppm utilizzando come riferimento il picco centrale del CDCl 3 Gli spettri infrarossi sono stati eseguiti con strumento NICOLET 5PC FT-IR SPECTROPHOTOMETER su film liquido nell'intervallo di frequenza 4000-400 cm-1.
I solventi e la maggior parte dei reagenti utilizzati sono quelli analitici commerciali. La dimetilformammide (DMF) e l'anisolo sono stati seccati su setacci molecolari 4Å.
In un palloncino codato da 25 ml si è pesato il palladio acetato e la base. Il palloncino è stato posto sotto vuoto e poi, dopo 15 minuti, sotto azoto. La soluzione ottenuta sciogliendo lo iodobenzene e il 2-norbornene in DMF è stata aggiunta agli altri reagenti nel pallone codato mettendo per ultimo l'olefina. La reazione è stata condotta per un tempo e ad una temperatura che verranno indicate di volta in volta. Dopo raffreddamento a temperatura ambiente il grezzo di reazione è stato ripreso con cloruro di metilene e trattato con acido solforico al 10%. La fase organica è stata separata dalla fase acquosa e seccata su solfato di sodio. Dopo filtrazione la soluzione ottenuta è stata messa sotto vuoto fino alla completa evaporazione del solvente, usando una trappola intermedia raffreddata in azoto liquido. Si è ripreso con poco cloruro di metilene e si è compiuta l'analisi gascromatografica. La separazione dei prodotti è stata eseguita mediante colonna cromatografica flash preparata con silice (ICN Silica 32-63, 60 Å), impaccando ed eluendo alla pressione di 0.9 bar. Gli eluenti usati sono miscele di esano e acetato di etile in proporzioni variabili a seconda dei prodotti da separare.
|
Reagenti |
||
|
Palladio acetato |
10.6 mg |
0.0473 mmole |
|
Carbonato di potassio |
70.0 mg |
0.5034 mmole |
|
Iodobenzene |
95.7 mg |
0.4691 mmole |
|
2-norbornene |
56.6 mg |
0.6021 mmole |
|
0.5 ml acrilato di metile |
||
|
1 ml DMF |
||
Procedimento e risultati
Si veda il procedimento generale.
La reazione è stata condotta a 80°C per 18h.
L'analisi GC ha messo in evidenza la formazione di due prodotti principali 15 e 16 ottenuti con rese gascromatografiche del 10% e del 20%, calcolate usando come standard interno il docosano e f = 1.10.
La conversione dello iodobenzene non è valutabile quantitativamente, in quanto lo iodobenzene non reagito viene allontanato totalmente dalla pompa meccanica durante il trattamento fatto per togliere l'acrilato di metile in eccesso, come accertato da prove in bianco.
Per la separazione si è eluito con esano:acetato di etile 9.5:0.5. L'eluizione è iniziata raccogliendo frazioni da 21 ml fino alla 13
a frazione; successivamente si sono raccolte frazioni da 7 ml.Il prodotto 15 è presente nelle frazioni 18-19; dalla frazione 20 alla frazione 21 i due prodotti sono usciti in miscela. Il prodotto 16 è nelle frazioni 22-23.
Si sono separati in totale 3 grezzi (preparati come sopra) per avere in tutto 11 mg di ciascuno dei due prodotti.
15 e 16 sono stati caratterizzati mediante
1H NMR, MS, IR.La reazione condotta a 80°C per 18h raddoppiando la quantità di acrilato di metile (1 ml) ha portato agli stessi risultati
Conducendo la reazione a 80°C per 7h la resa di 15 è stata del 7% mentre quella di 16 è stata del 9%.
Conducendo la reazione a 105°C per 18h, oltre a 15 (11%) e a 16 (8%), si formano altri due prodotti identificati attraverso GC-GM come 17 (4%) e cinnamato di metile (10%).
La reazione condotta a 55°C per 18h ha portato a 15 con resa del 4% e a 16 con resa del 5%.
Prodotto 15
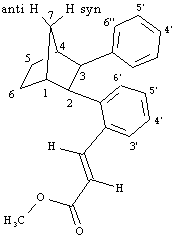
1H NMR (400 MHz):
7.97 (1H, d, J = 15.8 Hz, =CHAr), 7.33 (1H, dd, J = 7.9, 1.3 Hz, H6'), 7.22-7.15 (2H, m, H3', H5'), 6.98 (1H, td, J = 7.5, 1.4 Hz, H4'), 6.90-6.85 (3H, m, H3'', H4'', H5''), 6.84-6.80 (2H, m, H2'', H6''), 6.01 (1H, d, J = 15.8 Hz, =CHCO2Me), 3.84 (3H, s, OMe), 3.57 (1H, d, J = 9.7 Hz, H2), 3.32 (1H, dd, J = 9.7, 1.4 Hz, H3), 2.71 (1H, m, H1), 2.44 (1H, m, H4), 2.32 (1H, d di quintetti, J = 10.2, 2.0 Hz, H7syn), 1.88-1.69 (2H, m, H5exo, H6exo), 1.63-1.50 (3H, m, H5endo, H6endo, H7anti).
MS (70 eV):
M+ 332 (25), m/e 272 (30), 265 (25), 247 (15), 205 (55), 181 (45), 167 (25), 155 (40), 141 (95), 128 (50), 117 (65), 115 (100), 95 (20), 91 (90), 77 (20), 48 (20).
FT-IR: 1718, 1632.
Prodotto 16
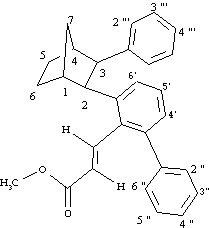
1H NMR (400 MHz):
7.66 (1H, d, J = 16.3 Hz, =CHAr), 7.34 (1H, dd, J = 8.0, 1.8 Hz, H6'), 7.26-7.19 (3H, m, H3'', H4'', H5''), 7.18 (1H, t, J = 7.8 Hz, H5'), 7.01-6.95 (3H, m, H3''', H4''', H5'''), 6.94 (1H, dd, J = 7.6, 1.2 Hz, H4') 6.92-6.87 (2H, m, H2''', H6''') 6.87-6.83 (2H, m, H2'', H6'') 5.20 (1H, d, J = 16.2 Hz, =CHCO2Me), 3.68 (3H, s, OMe), 3.49 (1H, d, J = 9.5 Hz, H2), 3.30 (1H, d, J = 9.5 Hz, H3), 2.74 (1H, m, H1), 2.44 (1H, m, H4), 2.35 (1H, d di quintetti, J = 10.3, 1.8 Hz, H7syn), 1.84-1.68 (2H, m, H5exo, H6exo), 1.60-1.40 (3H, m, H5endo, H6endo, H7anti)
MS (70 eV):
M+ 408 (55), m/e 257 (30), 217 (38), 191 (50), 117 (30), 91 (58), 73 (18), 52 (20), 48 (50)
FT-IR: 1718, 1636
|
Reagenti |
||
|
Palladio acetato |
10.0 mg |
0.0446 mmole |
|
Carbonato di potassio |
71.9 mg |
0.5388 mmole |
|
Iodobenzene |
91.1 mg |
0.4465 mmole |
|
2-norbornene |
74.9 mg |
0.7968 mmole |
|
1 ml stirene |
||
|
1ml DMF |
||
Procedimento e risultati
Si veda il procedimento generale.
La reazione è stata condotta a 80°C per 24h.
L'analisi gascromatografica ha messo in evidenza la formazione di 24 e di 25 come prodotti principali assieme ad altri numerosi prodotti presenti in piccole percentuali.
La resa gascromatografica di 24 (12%) e di 25 (24%) è stata calcolata usando come standard interno tetracosano e f = 1.05. Anche in questo caso non è stato possibile valutare quantitativamente lo iodobenzene reagito in quanto quest'ultimo viene allontanato durante il trattamento compiuto alla pompa meccanica per allontanare lo stirene in eccesso.
Per la separazione del grezzo di reazione si è utilizzato come eluente esano. Raccogliendo frazioni da 21 ml, il prodotto 24 è uscito alle frazioni 44-50.
Dopodiché si è cambiato eluente usando 200 ml di acetato di etile raccogliendo il tutto in un'unica frazione. Questa frazione è stata purificata su strato sottile di SiO
2 usando come eluente esano:acetato di etile 9.9:0.1 e ottenendo 25 puro.I prodotti 24 e 25 così purificati sono stati caratterizzati con
1 H NMR e con MS.La reazione condotta per 72h a 80°C ha portato ad una resa del 10% in 24 e a 25 con resa del 28%.
Prodotto 25
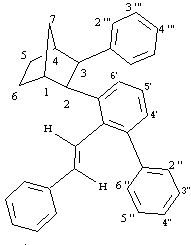
1
H NMR (400 MHz):7.34 (1H, d, H6'), 7.30 (1H, d, H2''' o H6'''), 7.25-7.09 (8H, m, 2H orto e 1H para dell'anello stirilico, 2'', 4'', 6'', 4''' e 5' centrato a 7.14), 7.04-6.88 (8H, m, 2H meta dell'anello stirilico, 3'', 5'', 5''', 3''', 4' centrato a 6.98, 6''' o 2''' centrato a 6.94), 6.80 (1H, d, J = 16.5 Hz, =CHAr), 5.88 (1H, d, J = 16.5 Hz, =CHPh), 3.62 (1H, d, J = 9.6 Hz, H2), 3.27 (1H, d, J = 9.6 Hz, H3), 2.75 (1H, m, H1), 2.43 (1H, m, H4), 2.37 (1H, d di quintetti, J = 10.1, 1.3 Hz, H7syn), 1.82-1.63 (2H, m, H5exo, H6exo), 1.61-1.41 (3H, m, H5endo, H6endo, H7anti).
MS (CI):
M+1 427
Prodotto 24:
MS (70 eV):
M
+ 350 (30), m/e 282 (20), 268 (15), 191 (20), 165 (13), 119 (14), 105 (21), 96 (27), 95 (43), 91 (50), 83 (42), 59 (61)
|
Reagenti |
||
|
Palladio acetato |
10.1 mg |
0.0451 mmole |
|
Acetato di potassio |
47.2 mg |
0.4816 mmole |
|
Iodobenzene |
91.3 mg |
0.4475 mmole |
|
2-norbornene |
52.6 mg |
0.5596 mmole |
|
1 ml acrilato di metile |
||
|
1 ml DMF |
Procedimento e risultati
Si veda procedimento generale.
La reazione è stata condotta a 105°C per 18h.
L'analisi gascromatografica ha mostrato la formazione di 17, di un prodotto non ancora ben caratterizzato di PM 350 e di cinnamato di metile (PM 162) assieme ad altri numerosi prodotti in piccole percentuali.
Il prodotto 17 e quello di PM 350 sono stati separati usando una miscela eluente esano:acetato di etile 9:1.
Si sono raccolte inizialmente 6 frazioni da 21 ml, poi frazioni da 7 ml. Il prodotto 17 è presente nelle frazioni 16 e 17. La frazione 18
a conteneva entrambi i prodotti. Nella frazione 19a è presente il prodotto di PM 350. Si sono separati quattro grezzi per ottenere 11 mg di ciascuno dei due prodotti.I prodotti sono stati caratterizzati con
1H NMR, GC-GM, FT-IR.A 80°C per 18h la reazione non avviene.
Prodotto 17
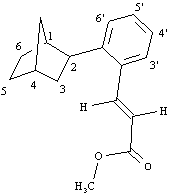
1H NMR (300MHz):
8.08 (1H, d, J = 15.8 Hz, =CHAr), 7.51 (1H, d, J = 7.6 Hz, H3'), 7.33-7.29 (2H, m, H5', H6'), 7.19-7.12 (1H, m, H4'), 6.29 (1H, d, J = 15.8 Hz, =CHCO2Me), 3.80 (3H, s, OMe), 2.98 (1H, dd, J = 9.1, 5.5 Hz, H2), 2.45 (1H, m, H1), 2.32 (1H, m, H4), 1.84 (1H, d, d, d, J = 12.0, 9.1, 2.3 Hz, H3endo), 1.70-1.58 (2H, m, H6exo, H5exo), 1.54 (1H, d con ulteriori accoppiamenti, J = 9.9 Hz, H7syn), 1.52-1.42 (1H, m, H3exo), 1.41-1.36 (1H, m, H6endo), 1.36-1.30 (1H, m, H5endo), 1.25 (1H, m, H7anti).
GC-GM (70 eV)
M
+ 256 (28), m/e 228 (25), 196 (28), 188 (28), 169 (55), 155 (25), 141 (42), 129 (100), 115 (65), 91 (15), 67 (20), 48 (15)FT-IR: 1719, 1631
Prodotto di PM 350
MS (70 eV):
M
+ 350 (45), m/e 255 (100) 195 (20), 165 (37), 152 (45), 141 (40), 129 (22), 115 (20), 95 (97), 79 (18), 67 (45), 48 (58)
|
Reagenti |
||
|
Palladio acetato |
10.0 mg |
0.0446 mmole |
|
Carbonato di potassio |
68.9 mg |
0.4957 mmole |
|
4-bromo-trifluorotoluene |
100.9 mg |
0.4484 mmole |
|
2-norbornene |
55,5 mg |
0.5904 mmole |
|
1 ml acrilato di metile |
||
|
1 ml DMF |
Procedimento e risultati
Si veda procedimento generale.
La reazione è stata condotta a 105°C per 18h.
L'analisi gascromatografica ha messo in evidenza il prodotto 1b e il prodotto 15b come prodotti principali assieme ad altri numerosi prodotti in piccola percentuale.
Per la separazione si è usato come eluente una miscela esano ed acetato di etile 9.5:0.5. Si sono raccolte frazioni da 21 ml. Il prodotto 1b è presente nelle frazioni 10-11, il prodotto 15b è nelle frazioni 24 -27.
I prodotti purificati sono stati caratterizzati mediante
1H NMR, MS e IR.Prodotto 1b
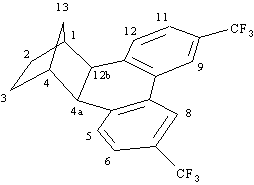
1
H NMR (300 MHz):8.04 (2H, s, H8, H9), 7.48 (2H, d, J = 8.1 Hz, H6, H11), 7.36 (2H, d, J = 8.1 Hz, H5, H12), 3.27 (2H, s, H4a, H12b), 2.40 (2H, m, H1, H4), 1.80-1.60 (4H, m, H2exo, H3exo, H2endo, H3endo), 1.32 (1H, d di quintetti, J = 10.2, 1.7 Hz, H13syn), 1.09 (1H, d di quintetti, J = 10.2, 1.5 Hz, H13anti).
MS (70 eV):
M
+ 382 (10), m/e 314 (80), 207 (30), 67 (100), 48 (95)Prodotto 15b
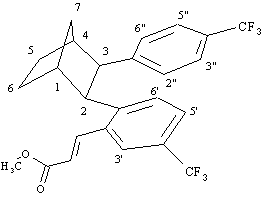
1
H NMR (300 MHz):7.91 (1H, d, J = 15.8 Hz, =CHAr), 7.43 (3H, s, H3', H5', H6'), 7.14 (2H, d, J = 8.2 Hz, H3", H5"), 6.90 (2H, d, J = 8.2 Hz, H2", H6"), 6.08 (1H, d, J = 15.8 Hz, =CHCO
2 Me), 3.85 (3H, s, OMe), 3.60 (1H, d, J = 9.7 Hz, H2), 3.39 (1H, d, J = 9.7 Hz, H3), 2.73 (1H, m, H1), 2.45 (1H, m, H4), 2.27 (1H, d di quintetti, J = 10.6, 1.9 Hz, H7syn), 1.91-1.69 (2H, m, H5exo, H6exo), 1.69-1.49 (3H, m, H7anti, H5endo, H6endo).MS (70 eV):
M
+ 468 (70), m/e 449 (100), 437 (50), 408 (70), 400 (45), 394 (50), 341 (30), 159 (15).FT-IR: 1720, 1631.
|
Reagenti |
||
|
Palladio acetato |
10.0 mg |
0.0446 mmole |
|
Carbonato di potassio |
63.7 mg |
0.4582 mmole |
|
4-bromo-trifluorotoluene |
103.2 mg |
0.4587 mmole |
|
2-norbornene |
62.8 mg |
0.6681 mmole |
|
2 ml DMF |
Procedimento e risultati
Si veda il procedimento generale.
La reazione è stata condotta a 105°C per 18h.
La fase organica analizzata in gascromatografia ha messo in evidenza la formazione di 1b e di 34 come prodotti principali assieme a piccole quantità di altri prodotti.
Per la separazione si è utilizzato esano come eluente raccogliendo frazioni da 21 ml. Il prodotto 1b è uscito nelle frazioni 15-19, mentre 34 è uscito nelle frazioni 22-23.
La resa in prodotto isolato di 1b è del 60% mentre quella di 34 è del 16%.
I prodotti purificati sono stati caratterizzati con
1H NMR, MS.Prodotto 34
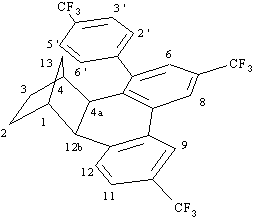
1
H NMR (300 MHz):8.12 (1H, m, H8), 8.10 (1H, m, H9), 7.74 (2H, d, J = 8.0 Hz, H3', H5'), 7.54-7.48 (3H, m, H11, H2', H6'), 7.37 (1H, d, J = 7.4 Hz, H12), 7.35 (1H, s, H6), 3.38 (1H, d, J = 9.9 Hz, H4a), 3.28 (1H, d, J = 9.9 Hz, H12b), 2.35 (1H, m, H1), 1.94 (1H, m, H4), 1.62 (1H, tt, J = 12.5, 4.3 Hz, H2exo), 1.52-1.40 (1H, m, H2endo), 1.30-1.28 (2H, m, H13syn, H3exo), 0.95 (1H, d di quintetti, J = 10.3, 1.4 Hz, H13anti), 0.80-0.69 (1H, m, H3endo).
MS (70 eV):
M
+ 526 (55), m/e 507 (15), 458 (100), 389 (40), 369 (30), 320 (55), 67 (75).
|
Reagenti |
||
|
Fenilnorbornilpalladio(II) cloruro |
9.9 mg |
0.0317 mmole |
|
Carbonato di potassio |
14.1 mg |
0.1014 mmole |
|
Iodobenzene |
13.2 mg |
0.0647 mmole |
|
2 ml DMF |
Procedimento e risultati
Un palloncino da 25 ml sotto azoto è stato tarato; sempre sotto azoto si sono pesati in successione nel pallone di reazione munito di ancoretta magnetica, il fenilnorbornil-palladio(II) cloruro e il carbonato di potassio.
Si è poi aggiunto lo iodobenzene sciolto in 2 ml di dimetilformammide dry. Si è fatto reagire sotto agitazione a temperatura ambiente per 24h.
Dopo tale periodo il palloncino è stato posto in corrente d'azoto ed è stato aggiunto sodio boroidruro in eccesso. Dopo avere lasciato reagire per 1 minuto per distruggere il complesso di palladio eventualmente ancora presente, si è aggiunto, goccia a goccia, al grezzo di reazione acido solforico al 5% fino alla scomparsa dell'effervescenza
La soluzione è stata ripresa con cloruro di metilene e trattata con acido solforico al 5%. La fase organica è stata estratta e seccata su solfato di sodio anidro. Dopo la filtrazione, la soluzione è stata analizzata alla GC-GM mettendo in evidenza la formazione di 1 come prodotto principale. Lo spettro di massa è identico a quello riportato in letteratura [1].
|
Reagenti |
||
|
Fenilnorbornilpalladio(II) cloruro |
9.9 mg |
0.0317 mmole |
|
Carbonato di potassio |
14.2 mg |
0.1021 mmole |
|
Iodobenzene |
12.6 mg |
0.0617 mmole |
|
2-norbornene |
10.9 mg |
0.1159 mmole |
|
2 ml DMF |
Procedimento e risultati
Si veda la reazione precedente.
La reazione è stata condotta a temperatura ambiente per 24h. Il 2-norbornene è stato sciolto con lo iodobenzene in DMF.
Il grezzo di reazione è stato analizzato alla GC-GM mettendo in evidenza la presenza dell'arilesaidrometanotrifenile 8, caratterizzato per confronto con i dati riportati in letteratura [4], insieme al prodotto di massa 248 probabilmente corrispondente al 2,3-difenilnorbornano.
|
Reagenti |
||
|
Pd(PPh 3)4 |
10.3 mg |
0.0089 mmole |
|
Sodio tetrafenilborato |
161.9 mg |
0.4733 mmole |
|
1-bromo-2-nitrobenzene |
94.5 mg |
0.4678 mmole |
|
2-norbornene |
104.3 mg |
0.1106 mmole |
|
2 ml anisolo |
Procedimento e risultati
Il palladio tetrakis e il sodio tetrafenilborato vengono pesati in un palloncino codato munito di ancoretta magnetica. Dopo 15 minuti di vuoto si aggiungono in atmosfera d'azoto l'1-bromo-2-nitrobenzene e il 2-norbornene sciolti in 2 ml di anisolo.
La reazione viene condotta sotto agitazione a 80°C per 36h e poi raffreddata a temperatura ambiente. Si riprende con cloruro di metilene e si tratta con acqua. Dopo filtrazione, l'analisi gascromatografica ha messo in evidenza la quasi totale conversione del 1-bromo-2-nitrobenzene e la formazione del 2-(2-nitrofenil)-3-fenilnorbornano e del 1-fenil-2-nitrobenzene come prodotto secondario.
La separazione dei prodotti è stata compiuta utilizzando una colonna cromatografica flash, impaccata con gel di silice, ed operando alla pressione di 0.9 bar. Si è eluito con una miscela esano:acetato di etile 9.5:0.5.
Raccogliendo frazioni da 21 ml il 2-(2-nitrofenil)-3-fenilnor-bornano, è uscito nelle frazioni 11-14 con resa in prodotto isolato del 40%, mentre il prodotto secondario, 2-fenil-nitrobenzene, è uscito nelle frazioni 17-19.
Il prodotto 2-(2-nitrofenil)-3-fenilnorbornano è stato caratterizzato con
1 H NMR, MS.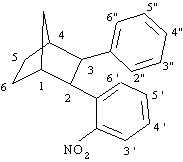
1
H NMR (300 MHz):7.61 (1H, dd, J = 8.0, 1.2 Hz, H3'), 7.34 (1H, dd, H6'), 7.30 (1H, td, H5'), 7.08 (1H , ddd, H4'), 6.93 (5H, m, H2", H3", H4", H5", H6"), 3.95 (1H, d, J = 9.6 Hz, H2 o H3), 3.49 (1H, d. J = 9.6 Hz, H3, H2), 2.60 (1H, m, H1 o H4), 2.55 (1H, m, H4 o H1), 2.34 (1H, d di quintetti, J = 10.3, 2.0 Hz, H7syn), 1.81-1.71 (2H, m, H2exo, H3exo), 1.61 (1H, d di quintetti, J = 10.3, 1.5 Hz, H7anti), 1.58-1.51 (2H, m, H2endo, H3endo).
MS (CI):
M+1
294
Procedimento
In un pallone da 50 ml a due colli si sono messi 600 mg di 2-(2-nitrofenil)-3-fenilnorbornano e un'ancoretta magnetica. Sul collo centrale si è montato un refrigerante a ricadere e sul collo laterale si è inserito un imbocco con rubinetto dal quale si è fatto flussare azoto. Dopo avere sciolto il prodotto in 8 ml di tetraidrofurano dry, si sono aggiunti 80 mg di palladio su carbone al 10% e la soluzione è stata raffreddata a 0°C con un bagno di ghiaccio e acqua.
Vengono quindi aggiunti 190 mg di sodio boroidruro in tre porzioni nel giro di 30 minuti dopodiché la soluzione è lasciata procedere a temperatura ambiente per 1h 40'. Alla fine si riprende con 50 ml di etere e si filtra. Dopo filtrazione si distrugge l'eccesso di sodio boroidruro aggiungendo 10 ml di acqua e poche gocce di HCl 2N, avendo cura di non scendere al di sotto di pH 6. Dopo avere separato la fase organica e averla seccata su solfato di sodio, è stata fatta un'analisi gascromatografica che ha messo in evidenza due prodotti: uno corrispondente al nitroderivato non reagito e l'altro successivamente caratterizzato come il 2-(2-aminofenil)-3-fenilnorbornano. Per separare i due prodotti si è impiegata una colonna cromatografica flash impaccata con gel di silice eluendo con una miscela esano:acetato di etile 9.5:0.5 fino alla 27a frazione. Dopodiché si è usato come eluente esano:acetato di etile 9:1. Raccogliendo frazioni da 21 ml il nitroderivato è uscito alle frazioni 11-14 e se ne sono recuperati 300 mg. Il 2-(2-aminofenil)-3-fenilnorbornano è uscito alle frazioni 27-39 e se ne sono raccolti 100 mg con resa del 20%.
Il prodotto della riduzione è stato caratterizzato con 1H NMR, 13C NMR, GC-GM.
2-(2-aminofenil)-3-fenilnorbornano:
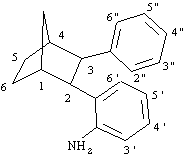
1
H NMR (300 MHz):7.30 (1H, dd, J = 7.7, 1.4 Hz, H6'), 7.08-6.96 (5H, m, protoni sull'anello fenilico non sostituito), 6.90 (1H, td, J = 7.5, 1.5 Hz, H4'), 6.75 (1H, td, J = 7.5, 1.3 Hz, H5'), 6.36 (1H, dd, J = 7.8, 1.3 Hz, H3'), 3.29 (1H, d, J = 9.25 Hz, H2), 3.26 (2H, m, NH
2), 3.19 (1H, d, J = 9.5 Hz, H3), 2.80 (1H, m, H1), 2.42 (1H, m, H4), 2.35 (1H, d di quintetti, J = 10.1, 2.0 Hz, H7syn), 1.91-1.70 (2H, m, H5exo, H6exo), 1.62-1.50 (3H, m, H5endo, H6endo, H7anti).13
C NMR (75.47 MHz):143.8 Cq, 142.7 Cq, 128.5, 128.1 Cq, 127.1, 126.4 C6', 126.3 C4', 125.5, 118.4 C5', 115.6 C3', 54.8 C3, 49.1 C2, 45.1 C4, 40.3 C1, 36.5 C7, 31.1, 30.4 C5, C6
GC-GM (70 eV):
M
+ 263 (50), m/e 172 (25), 132 (75), 106 (100), 91 (30)
[1] M. Catellani, G.P. Chiusoli, J. Organomet. Chem., 286 (1985) C13.
[2] M.C. Gallazzi, T.L. Hanlon, G. Vitulli, L. Porri, J. Organomet.Chem., 33 (1975) 90.
[3] J. Sicher, Angew. Chem. Int. Ed. Engl., 11 (1972) 200.
[4] O. Reiser, M. Weber, A. de Meijere, Angew. Chem. Int. Ed. Engl., 28 (1989) 1037; K. Albrecht, O. Reiser, M. Weber, B. Knieriem, A. de Meijere, Tetrahedron, 50 (1994) 383.
[5] R.F. Heck, "Palladium Reagents in Organic Synthesis", Acad. Press, London, 1985.
[6] M. Catellani, M.C. Fagnola, Angew. Chem. Int. Ed Engl., 33 (1994) 2421; F. Frignani, Tesi di Laurea, 1995, Università di Parma.
[7] G. Bocelli, M. Catellani, S. Ghelli, J. Organomet. Chem., 458 (1993) C12.
[8] M. Catellani, M.C. Fagnola, B. Marmiroli, D. Acquotti, J. Organomet. Chem., in corso di stampa.
[9] M.A. Bennett, H.P. Schwemlein, Angew. Chem. Int. Ed. Engl., 28 (1989) 1296.
[10] M. Catellani, G.P. Chiusoli, S. Concari, Tetrahedron, 45 (1989) 5263.
[11] M. Petrini, R. Ballini, G. Rosini, Synthesis (1987) 713.
Indice
|
Impostazione della tesi |
1 |
|
|
Introduzione |
3 |
|
|
Risultati e discussione |
8 |
|
|
Parte sperimentale |
25 |
|
|
Bibliografia |
54 |